Using the Python API
This tutorial shows how to use the MS²Rescore Python API for each step of the rescoring process individually. This is useful if you want to customize rescoring for your own Python workflow or if you want to understand how MS²Rescore works.
Note that the full MS²Rescore workflow is also available from Python with the single function call ms2rescore.rescore().
[1]:
import logging
import plotly.io
logging.basicConfig(level=logging.INFO)
plotly.io.renderers.default = "plotly_mimetype+notebook"
Reading and parsing peptide-spectrum matches
[2]:
from psm_utils.io import read_file
from ms2rescore.report.charts import score_histogram
INFO:numexpr.utils:Note: NumExpr detected 32 cores but "NUMEXPR_MAX_THREADS" not set, so enforcing safe limit of 8.
INFO:numexpr.utils:NumExpr defaulting to 8 threads.
Reading the PSM file
MS²Rescore is fully centered around the use of a psm_utils PSMList. This is a unified data representation of PSMs and their various attributes. Internally, it is simply a list of Pydantic data classes which represent PSMs. With the submodule psm_utils.io, we can read PSMs from a variety of file formats. Here, we will read a PSM file in the MaxQuant msms.txt format.
Importantly, for rescoring, the PSM file must contain all target and decoy PSMs, including PSMs that did not pass the FDR threshold. Most search engines must be specifically configured to return all PSMs without FDR filtering.
[3]:
psm_list = read_file("../../../examples/id/msms.txt", filetype="msms")
psm_list["spectrum_id"] = [str(spec_id) for spec_id in psm_list["spectrum_id"]]
For a quick inspection, we can format the PSM list as a Pandas dataframe and display the first few rows:
[4]:
psm_list.to_dataframe().head()
[4]:
| peptidoform | spectrum_id | run | collection | spectrum | is_decoy | score | qvalue | pep | precursor_mz | retention_time | ion_mobility | protein_list | rank | source | provenance_data | metadata | rescoring_features | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | AAAAAAALQAK/2 | 4703 | 20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02 | None | None | False | 107.660 | None | 0.001517 | 478.77982 | 5.2007 | None | [P36578, H3BM89, H3BU31] | None | msms | {'msms_filename': '..\..\..\examples\id\msms.t... | {'Scan index': '3698', 'Sequence': 'AAAAAAALQA... | {} |
| 1 | [ac]-AAAAAEQQQFYLLLGNLLSPDNVVR/3 | 13572 | 20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02 | None | None | False | 107.740 | None | 0.004931 | 915.15197 | 11.8470 | None | [O00410, E7ETV3, E7EQT5, C9JZD8] | None | msms | {'msms_filename': '..\..\..\examples\id\msms.t... | {'Scan index': '11885', 'Sequence': 'AAAAAEQQQ... | {} |
| 2 | [ac]-AAAAAEQQQFYLLLGNLLSPDNVVRK/3 | 13366 | 20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02 | None | None | False | 137.890 | None | 0.000493 | 957.85029 | 11.6900 | None | [O00410, E7ETV3, E7EQT5, C9JZD8] | None | msms | {'msms_filename': '..\..\..\examples\id\msms.t... | {'Scan index': '11695', 'Sequence': 'AAAAAEQQQ... | {} |
| 3 | AAAAAQGGGGGEPR/2 | 505 | 20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02 | None | None | False | 22.641 | None | 0.142020 | 585.28653 | 0.5178 | None | [E9PJF0, E9PQW4, P27361] | None | msms | {'msms_filename': '..\..\..\examples\id\msms.t... | {'Scan index': '419', 'Sequence': 'AAAAAQGGGGG... | {} |
| 4 | AAAAAWEEPSSGN[de]GTAR/2 | 6589 | 20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02 | None | None | False | 89.403 | None | 0.046504 | 823.87389 | 6.6105 | None | [Q9P258] | None | msms | {'msms_filename': '..\..\..\examples\id\msms.t... | {'Scan index': '5439', 'Sequence': 'AAAAAWEEPS... | {} |
We can also directly plot the current PSM score distributions:
[5]:
score_histogram(psm_list)
Parsing modification names
While psm_utils could take care of all file parsing, we must still map the amino acid modification names that were used by the search engine to ones that are recognized by tools such as MS²PiP and DeepLC. This includes:
Names as used in the Unimod or PSI-MOD databases
Accession numbers as used in the Unimod or PSI-MOD databases
Chemical formulas
Mass shifts in Da
Note that, for instance DeepLC, requires a chemical formula to encode modifications. It will not be able to correctly encode modifications if only the mass shift is provided. It is therefore always preferred to provide a name/accession of a database where this information can be retrieved, or provide the chemical formula directly.
If a chemical formula is provided, other tools, such as MS²PIP, can use it to derive the correct mass shift.
To map modification names, simply provide a dictionary to the psm_list.map_modifications method.
[6]:
psm_list.rename_modifications({
"gl": "Gln->pyro-Glu",
"ox": "Oxidation",
"ac": "Acetylation",
"de": "Deamidation",
})
Assigning fixed modifications
Some search engines (although not many) do not report fixed (sometimes also called static) modifications in their output PSM files. This is for instance the case for MaxQuant. With the method psm_list.add_fixed_modifications, we can systematically assign fixed modifications to their amino acid targets, as was done during the search.
Note that add_fixed_modifications adds the modification in the ProForma 2.0 encoding as a prefix. To fully apply the modifications in the sequence, use the psm_list.apply_fixed_modifications method. A PSM ACDE/2 would therefore go from <[U:4]@C>ACDE/2 to AC[U:4]DE/2.
[7]:
psm_list.add_fixed_modifications([("U:Carbamidomethyl", ["C"])])
psm_list.apply_fixed_modifications()
Adding rescoring features
[8]:
import pandas as pd
from ms2rescore.feature_generators.basic import BasicFeatureGenerator
from ms2rescore.feature_generators.maxquant import MaxQuantFeatureGenerator
from ms2rescore.feature_generators.ms2pip import MS2PIPFeatureGenerator
from ms2rescore.feature_generators.deeplc import DeepLCFeatureGenerator
The PSM list, as read from a MaxQuant msms.txt file, currently does not contain any rescoring features:
[9]:
pd.DataFrame(list(psm_list["rescoring_features"]))
[9]:
| 0 |
|---|
| 1 |
| 2 |
| 3 |
| 4 |
| ... |
| 35551 |
| 35552 |
| 35553 |
| 35554 |
| 35555 |
35556 rows × 0 columns
Note that psm_list[rescoring_features] returns a Numpy array of dictionaries, but we can use Pandas to format it as a table.
Basic features
Basic features from BasicFeatureGenerator can be added from any PSM list and contain simple feature, such as the search engine score, charge state, and absolute precursor mass error.
[10]:
basic_fgen = BasicFeatureGenerator()
basic_fgen.add_features(psm_list)
INFO:ms2rescore.feature_generators.basic:Adding basic features to PSMs.
[11]:
pd.DataFrame(list(psm_list["rescoring_features"]))
[11]:
| charge_n | charge_1 | charge_2 | charge_3 | charge_4 | charge_5 | charge_6 | charge_7 | abs_ms1_error_ppm | search_engine_score | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1.137810 | 107.6600 |
| 1 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.598941 | 107.7400 |
| 2 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.573291 | 137.8900 |
| 3 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.929236 | 22.6410 |
| 4 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.666129 | 89.4030 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 35551 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.782088 | 76.1000 |
| 35552 | 4 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1.147761 | 114.6400 |
| 35553 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.813101 | 43.1240 |
| 35554 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.719097 | 23.5620 |
| 35555 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1.083807 | 9.1885 |
35556 rows × 10 columns
Now the PSMs contain a simple set of features.
Note that the charge state is present as both an integer and a one-hot encoded vector. This is because charge state can act as both a categorical and a numerical feature. Simply put, sometimes an effect increases or decreases with increasing charge state, and sometimes it is simply different for different charge states.
MS²PIP features
MS²PIP is a machine learning tool that predicts the MS2 spectrum of a peptide given its sequence. It is previously identified MS2 spectra and their corresponding peptide sequences. Because MS²PIP uses the highly performant - but traditional - machine learning approach XGBoost, it can already produce accurate predictions even if trained on smaller spectral libraries. This makes MS²PIP a very flexible platform to train new models on custom datasets. Nevertheless, MS²PIP comes with several pre-trained models, which we will use in this tutorial.
Because traditional proteomics search engines do not fully consider MS2 peak intensities in their scoring functions, adding rescoring features derived from spectrum prediction tools has proved to be a very effective way to further improve the sensitivity of peptide-spectrum matching. Generating features from MS²PIP predictions follows these steps:
Predict MS2 spectra for all PSMs in the dataset, including decoy PSMs and low scoring target PSMs.
Read and parse the observed MS2 spectra from the original spectrum files (MGF or mzML).
Compare the predicted and observed spectra using various similarity metrics, which are returned as rescoring features.
Configuring MS²PIP
In contrast to the basic feature generator, MS²PIP requires some parameters to be set and requires access to the original observed peptide spectra:
model: Name of the prediction model to be used. This strongly depends on the dataset your are rescoring. A list of all MS²PIP models is available on https://ms2pip.readthedocs.io/en/latest/prediction-models/.ms2_tolerance: As MS²PIP must reannotate the observed MS2 spectra, a mass tolerance must be set. For MS²PIP, this is configured in Dalton. A good value for Orbitrap spectra, for example, is0.02.spectrum_path: Path to the original spectrum files. This can be a single file, or a directory containing multiple files. The spectrum files must be in MGF or mzML format.spectrum_id_pattern: A regular expression pattern to extract the spectrum ID from the PSMs (see below).processes: Number of CPU processes to use for parallel processing. Note that higher values can lead to memory issues.
[12]:
ms2pip_fgen = MS2PIPFeatureGenerator(
model="HCD",
ms2_tolerance=0.02,
spectrum_path="../../../examples/mgf/20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02.mgf",
spectrum_id_pattern=r".*scan=(\d+)$",
processes=8,
)
A very import aspect of generating MS²PIP-derived features is making sure the peptide identifications are matched to the correct spectra. Unfortunately, many search engines alter spectrum identifiers in their output files. In the case of MaxQuant and data from Thermo instruments, fortunately, the scan numbers are returned. If the spectrum files have been converted to MGF with ThermoRawFileParser, we need to extract these exact scan numbers from the spectrum TITLE entries. This can be done
with a regular expression pattern. For example, the following pattern extracts the scan number from the following spectrum title:
spectrum_id_pattern = r"scan=(\d+)"
mzspec=20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02.raw: controllerType=0 controllerNumber=1 scan=2
We can test this with the first spectrum in the file:
[13]:
from pyteomics.mgf import read as read_mgf
for spectrum in read_mgf("../../../examples/mgf/20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02.mgf"):
print(spectrum["params"]["title"]) # noqa T201
break
mzspec=20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02.raw: controllerType=0 controllerNumber=1 scan=2
[14]:
import re
spectrum_id = re.match(r".*scan=(\d+)$", spectrum["params"]["title"]).group(1)
print(spectrum_id) # noqa T201
2
We can use this scan number to retrieve its corresponding PSM:
[15]:
psm_list[psm_list["spectrum_id"] == spectrum_id]
[15]:
PSMList(
psm_list=[
PSM(
peptidoform=Peptidoform('ELMKLAPR/2'),
spectrum_id='2',
run='20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02',
collection=None,
spectrum=None,
is_decoy=False,
score=58.194,
qvalue=None,
pep=0.11486,
precursor_mz=479.28114,
retention_time=0.0054151,
ion_mobility=None,
protein_list=['K7ER25', 'K7EJY1', 'Q7Z3S7', 'E7EUE0'],
rank=None,
source='msms',
provenance_data={
'msms_filename': '..\\..\\..\\examples\\id\\msms.txt'
},
metadata={
'Scan index': '0',
'Sequence': 'ELMKLAPR',
'Length': '8',
'Missed cleavages': '1',
'Modifications': 'Unmodified',
... +51
},
rescoring_features={
'charge_n': 2,
'charge_1': 0,
'charge_2': 1,
'charge_3': 0,
'charge_4': 0,
... +5
}
)
]
)
Generating MS²PIP features
[16]:
ms2pip_fgen.add_features(psm_list)
INFO:ms2rescore.feature_generators.ms2pip:Adding MS²PIP-derived features to PSMs.
INFO:ms2rescore.feature_generators.ms2pip:Running MS²PIP for PSMs from run `20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02`...
INFO:ms2pip.core:Processing spectra and peptides...
WARNING:ms2rescore.feature_generators.ms2pip:Failed to calculate features for 2 PSMs
[17]:
pd.DataFrame(list(psm_list["rescoring_features"]))
[17]:
| charge_n | charge_1 | charge_2 | charge_3 | charge_4 | charge_5 | charge_6 | charge_7 | abs_ms1_error_ppm | search_engine_score | ... | iony_abs_diff_Q2 | iony_abs_diff_Q3 | iony_mean_abs_diff | iony_std_abs_diff | dotprod | dotprod_ionb | dotprod_iony | cos | cos_ionb | cos_iony | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1.137810 | 107.6600 | ... | 0.004537 | 0.009003 | 0.005573 | 0.004217 | 0.007582 | 6.011568e-03 | 0.001571 | 0.965756 | 0.986066 | 0.948538 |
| 1 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.598941 | 107.7400 | ... | 0.001146 | 0.003543 | 0.003072 | 0.004661 | 0.013845 | 8.715502e-03 | 0.005130 | 0.950527 | 0.972807 | 0.984188 |
| 2 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.573291 | 137.8900 | ... | 0.001195 | 0.002719 | 0.001657 | 0.001813 | 0.010731 | 9.636219e-03 | 0.001095 | 0.930393 | 0.933799 | 0.973310 |
| 3 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.929236 | 22.6410 | ... | 0.006460 | 0.015503 | 0.012758 | 0.016877 | 0.002545 | 1.657421e-03 | 0.000887 | 0.626743 | 0.900722 | 0.415943 |
| 4 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.666129 | 89.4030 | ... | 0.001901 | 0.011982 | 0.010727 | 0.019760 | 0.001069 | 3.751754e-04 | 0.000694 | 0.720658 | 0.976536 | 0.662783 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 35551 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.782088 | 76.1000 | ... | 0.016826 | 0.030917 | 0.019391 | 0.015875 | 0.002695 | 1.119519e-03 | 0.001575 | 0.839569 | 0.991228 | 0.776745 |
| 35552 | 4 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1.147761 | 114.6400 | ... | 0.000678 | 0.002492 | 0.001421 | 0.001660 | 0.001290 | 4.580596e-04 | 0.000832 | 0.852407 | 0.987883 | 0.977172 |
| 35553 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.813101 | 43.1240 | ... | 0.014101 | 0.016411 | 0.015302 | 0.016832 | 0.001289 | 1.163584e-03 | 0.000125 | 0.386544 | 0.886590 | 0.076230 |
| 35554 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.719097 | 23.5620 | ... | 0.003215 | 0.007025 | 0.005133 | 0.006286 | 0.000170 | 1.747118e-07 | 0.000170 | 0.207400 | 0.007175 | 0.248114 |
| 35555 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1.083807 | 9.1885 | ... | 0.000841 | 0.002671 | 0.003405 | 0.004654 | 0.000018 | 3.528620e-12 | 0.000018 | 0.073865 | 0.767783 | 0.233401 |
35556 rows × 81 columns
Generating DeepLC features
[18]:
deeplc_fgen = DeepLCFeatureGenerator(
lower_score_is_better=False,
calibration_set_size=0.15,
spectrum_path=None,
processes=8,
deeplc_retrain=False,
)
deeplc_fgen.add_features(psm_list)
INFO:ms2rescore.feature_generators.deeplc:Adding DeepLC-derived features to PSMs.
INFO:ms2rescore.feature_generators.deeplc:Running DeepLC for PSMs from run (1/1): `20161213_NGHF_DBJ_SA_Exp3A_HeLa_1ug_7min_15000_02`...
Generating features from MaxQuant output
While MaxQuant msms.txt files do not contain directly usable rescoring features, it does contain various metrics from spectrum matching from which we can calculate rescoring features. These include features related to the mass error of the annotated MS2 peaks and the summed intensity of the annotated peaks.
The data that is used to calculate these features is stored in the PSM metadata attributes. For instance:
[19]:
pd.DataFrame(list(psm_list["metadata"]))[[
"Matches", "Intensities", "Mass Deviations [Da]", "Intensity coverage"
]].head()
[19]:
| Matches | Intensities | Mass Deviations [Da] | Intensity coverage | |
|---|---|---|---|---|
| 0 | y1;y2;y3;y4;y5;y6;y7;y8;y9;y1-NH3;y2-NH3;y3-NH... | 6420431.2;5259286.7;7595262.2;8731264.7;116766... | -9.10020734181671E-05;-0.000929981532976853;-0... | 0.450133939639015 |
| 1 | y1;y2;y4;y6;y7;y8;y9;y10;y11;y12;y13;y14;y2-NH... | 357644.7;48235;495609.6;1150605.6;1479450.3;14... | -0.000377184504145589;-0.000336244570064537;-0... | 0.682793460933943 |
| 2 | y1;y2;y3;y4;y5;y6;y7;y8;y9;y10;y11;y12;y13;y14... | 467523.3;1624841.4;1272199.7;504549;6773740.4;... | -0.000650374560791533;0.000150255794096665;0.0... | 0.632290650531871 |
| 3 | y2;y5;y5-H2O;a2;b2;b3 | 245612.6;40630.3;18142.2;108568;152454.4;155335.3 | -0.0009548283943559;0.00245136965645543;0.0023... | 0.187275289286221 |
| 4 | y1;y4;y6;y9;y10;y11;y12;y13;y11-H2O;y1-NH3;a2;... | 9529767.6;1651203.2;270327.2;11359473.6;212221... | -0.000178087084577783;0.00014824673877456;0.00... | 0.0479428579117575 |
[20]:
maxquant_fgen = MaxQuantFeatureGenerator()
maxquant_fgen.add_features(psm_list)
INFO:ms2rescore.feature_generators.maxquant:Adding MaxQuant-derived features to PSMs.
Assessing individual feature performance before rescoring
[21]:
from ms2rescore.report.charts import (
calculate_feature_qvalues,
feature_ecdf_auc_bar,
fdr_plot,
ms2pip_correlation,
)
import deeplc.plot
features = pd.DataFrame(list(psm_list["rescoring_features"]))
The following plot shows the number of identified PSM at varying FDR thresholds, where the red line indicates the commonly used 1% FDR threshold.
[22]:
psm_list.calculate_qvalues() # msms.txt does not contain q-values
fig = fdr_plot(psm_list.to_dataframe(), fdr_thresholds=None, log=False)
fig.show()
We can make a similar plot for each of the rescoring features by simply calculating q-values as if the feature were a search engine score.
[23]:
feature_qvalues, feature_ecdf_auc = calculate_feature_qvalues(
features=features,
is_decoy=psm_list["is_decoy"],
)
[24]:
feature_names = {
"basic": basic_fgen.feature_names,
"ms2pip": ms2pip_fgen.feature_names,
"deeplc": deeplc_fgen.feature_names,
"maxquant": maxquant_fgen.feature_names,
}
feature_generator_map = {name: gen for gen, f_list in feature_names.items() for name in f_list}
feature_qvalues_melt = feature_qvalues.melt(var_name="feature", value_name="q-value")
feature_qvalues_melt["feature_generator"] = feature_qvalues_melt["feature"].map(feature_generator_map)
[25]:
# Plot takes 100MB in saved file...
# fig = px.ecdf(
# data_frame=feature_qvalues_melt,
# x="q-value",
# color="feature_generator",
# line_dash="feature",
# )
# fig.show()
The larger the area under the curve (AUC) is for each line, the better. We can therefore visualize the same information in a more easy-to-interpret bar chart with the AUCs, which were already calculated by the calculate_feature_qvalues function.
[26]:
feature_ecdf_auc["feature_generator"] = feature_ecdf_auc["feature"].map(feature_generator_map)
[27]:
fig = feature_ecdf_auc_bar(feature_ecdf_auc)
fig.show()
Importantly, a low q-value ECDF AUC does not necessarily mean that a feature is not valuable for rescoring. The more orthogonal a feature is to the other information already used in PSM scoring, the more it will help to confidently identify PSMs. For instance, retention time features are completely independent from the information used in a traditional search engine. When combined with other scoring information, they can provide a substantial boost in sensitivity.
The quality of specific rescoring features can also be assessed in a manner specific to the feature. For instance, we can plot the distribution of the MS²PIP pearson correlations for target PSMs that initially passed the FDR threshold:
[28]:
fig = ms2pip_correlation(
features=features,
is_decoy=psm_list["is_decoy"],
qvalue=psm_list["qvalue"]
)
fig.show()
For DeepLC, we can plot the predicted retention time versus the observed retention time:
[29]:
fig = deeplc.plot.scatter(
df=features[(~psm_list["is_decoy"]) & (psm_list["qvalue"] <= 0.01)], # noqa: E712
predicted_column="predicted_retention_time_best",
observed_column="observed_retention_time_best",
)
fig.show()
DeepLC also provides a function to plot the current relative mean absolute error (rMAE) of the predicted retention times against a distribution of 460 benchmark datasets. The lower the rMAE, the better.
[30]:
fig = deeplc.plot.distribution_baseline(
df=features[(~psm_list["is_decoy"]) & (psm_list["qvalue"] <= 0.01)], # noqa: E712
predicted_column="predicted_retention_time_best",
observed_column="observed_retention_time_best",
)
fig.show()
Rescoring
Using Mokapot directly
[31]:
import matplotlib.pyplot as plt
from mokapot import brew
from ms2rescore.rescoring_engines.mokapot import convert_psm_list
%matplotlib inline
In this tutorial, we will use Mokapot as rescoring engine. By default it employs the same methodologies as Percolator, but is fully implemented in Python. First, the PSM list must be converted to a Mokapot LinearPsmDataset.
[32]:
linear_psm_dataset = convert_psm_list(psm_list)
Then, the Mokapot brew function can be called to rescore the PSMs:
[33]:
confidence_results, models = brew(linear_psm_dataset)
Mokapot contains a function to conveniently plot the results:
[34]:
confidence_results.plot_qvalues()
plt.show()
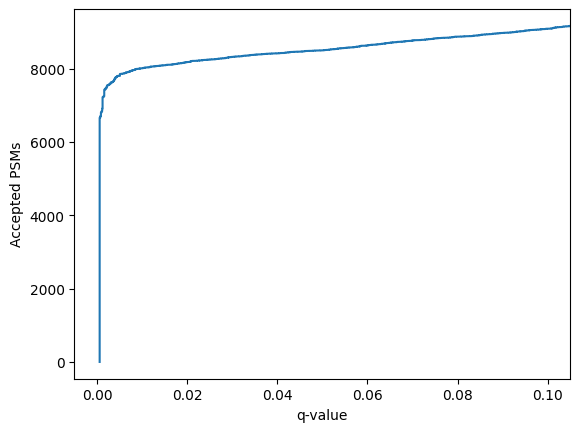
Or to compare the un-rescored PSMs to the rescored PSMs:
[35]:
linear_psm_dataset.assign_confidence().plot_qvalues(label="Before rescoring")
confidence_results.plot_qvalues(label="After rescoring")
plt.legend()
plt.show()
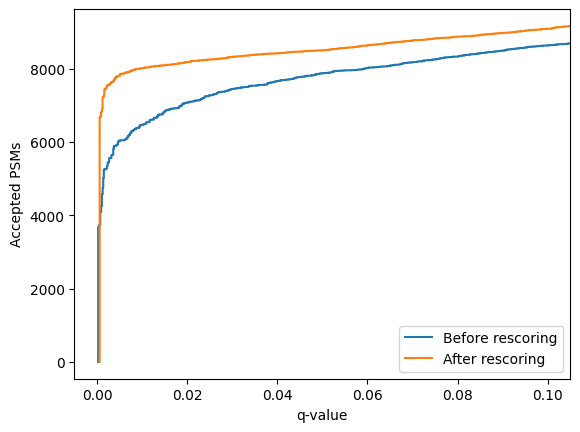
Using mokapot directly allows us to quickly generate LinearPsmDataset objects with different feature sets, for instance:
Only basic features
Basic + MaxQuant-derived features (mimicking a traditional Percolator rescoring run)
Basic + MaxQuant-derived + MS²PIP features
Basic + MaxQuant-derived + MS²PIP + DeepLC features
[36]:
feature_sets = {
"basic": feature_names["basic"],
"basic_mq": feature_names["basic"] + feature_names["maxquant"],
"basic_mq_ms2pip": feature_names["basic"] + feature_names["maxquant"] + feature_names["ms2pip"],
"basic_mq_ms2pip_deeplc": feature_names["basic"] + feature_names["maxquant"] + feature_names["ms2pip"] + feature_names["deeplc"],
}
[37]:
linear_psm_dataset.assign_confidence().plot_qvalues(label="Before rescoring")
for feature_set, features in feature_sets.items():
linear_psm_dataset = convert_psm_list(psm_list, features)
confidence_results, models = brew(linear_psm_dataset)
confidence_results.plot_qvalues(label=f"Rescoring with: {feature_set}")
plt.xscale('log')
plt.xlim(1e-4, 1)
plt.legend()
plt.show()
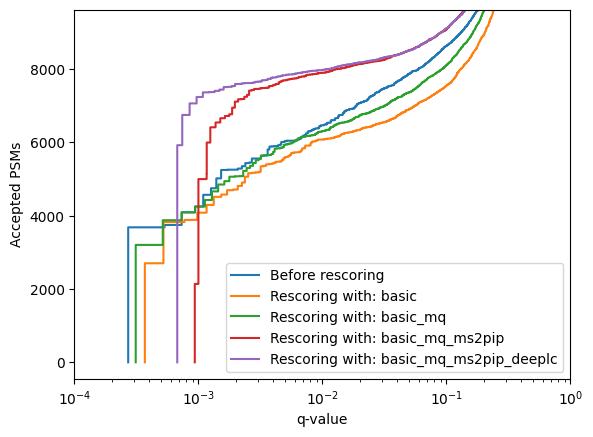
Using Mokapot within MS²Rescore
Mokapot can also be used for rescoring with the higher-level integration in MS²Rescore. Simply pass a PSMList object with rescoring features added to the rescore function. Doing so will automatically convert the PSMList to a Mokapot LinearPsmDataset, run the brew function, and update the original PSMList with the new scores, q-values and PEPs.
[38]:
from ms2rescore.rescoring_engines import mokapot
mokapot.rescore(psm_list)
[39]:
score_histogram(psm_list.to_dataframe())